1. 生物等效性#
在本节中我们将介绍生物等效性(bioequivalence)研究的基本理论知识,包括生物等效性的定义、常见的交叉试验设计方案、生物等效性研究设计要点以及相关的数据分析方法等。
若想获取交叉设计平均生物等效性分析的用户界面使用方法以及结果表格解读,可移步至:交叉设计 ABE 分析。
若想获知生物等效性相关的算法理论,可移步至:生物等效性算法简析。
1.1. 生物等效性的定义#
生物等效性是指在相似的试验条件下单次或多次给予相同剂量的试验药物后,受试制剂中药物的吸收速度和吸收程度与参比制剂的差异在可接受范围内。
生物等效性研究则是以药代动力学参数为终点指标,分析受试制剂与参比制剂是否具有生物等效性的人体试验。生物等效性是仿制药评价的重要指标:若仿制药与参比药具有生物等效性,则可以认为两者临床疗效相当——无需额外的临床试验即可获得上市批准。常被比较的药代动力学参数包括血药浓度-时间曲线下面积(AUC)、峰浓度(Cmax)等。此外,需要注意的是,生物等效性研究不关注临床疗效终点,因此生物等效性与临床等效性(clinical equivalence)略有不同。
生物等效性研究中评价生物等效性最常用的统计方法为比较平均生物等效性(average bioequivalence,ABE),其比较的是药代动力学参数的平均水平。一般而言,当受试制剂与参比制剂的药代动力学参数几何均值比(geometric mean ratio,GMR)在 80% 至 125% 间即可认为两者生物等效。
此外,对于一些喷雾剂、注射剂、脂质体以及其他复杂制剂则可以采用群体生物等效性(population bioequivalence,PBE)或个体生物等效性(individual bioequivalence,IBE)的方法。PBE 考察的是两个制剂间药代动力学参数的分布特征是否相同;而 IBE 则进一步要求两个制剂的药代动力学参数在不同个体内应当接近,考虑了个体内变异及个体与制剂的交互作用。
1.2. 交叉试验设计#
生物等效性研究一般的试验设计方法包括:
两制剂、单次给药、非重复交叉试验设计:最常用的试验设计方法,即受试者按随机顺序依次接受受试制剂与参比制剂。
两制剂、单次给药、平行试验设计:两种制剂分别在两组受试者中进行试验,适合于半衰期较长的药物。
两制剂、重复交叉试验设计:适合于高变异药物,受试者将重复接受某一制剂,使用此方法可以减少需要入组的受试者数量。
上文所说的交叉试验设计(crossover design)是一种将自身对照和组间比较相结合的设计方法。在交叉设计的临床试验中,同一个受试者在试验的不同周期将分别接受不同的制剂,由此可以评价不同治疗方案的差异,且相较于平行设计具有更高的试验效率。
以常见的 2x2 交叉试验设计为例,假设制剂分别为 A 药与 B 药。受试者将被随机的分为两组,其中第一组在周期 1 接受 A 药、周期 2 接受 B 药,其接受药物的顺序(一般称为序列,sequence)为 A-B;第二组则恰好相反,其序列为 B-A。在给药周期之间,需要设置足够长的洗脱期(wash-out period)以保证后一周期的临床结果不受前一周期的药物效应影响。示例可见 图 1.38。
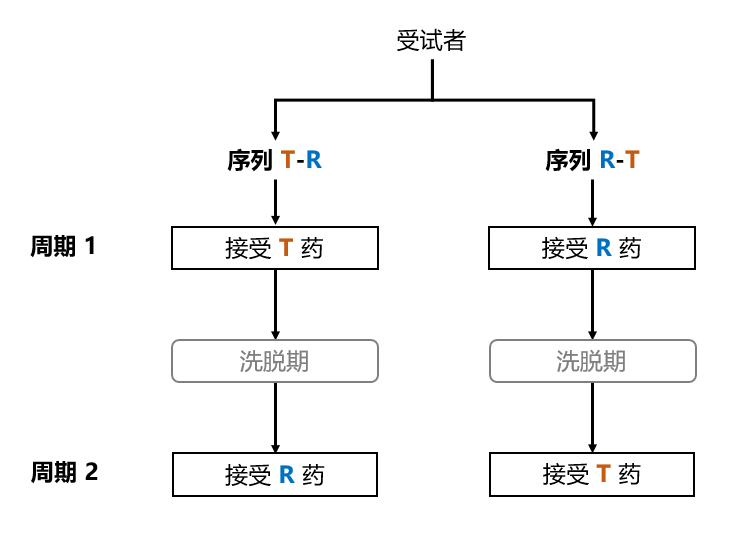
图 1.38 2x2 交叉试验设计示意图#
当存在周期与药物的交互效应或需要分析 PBE 与 IBE 时,2x2 交叉试验设计的结果无法提供足够的信息,因此需要进行多周期的试验或重复给药。常见的重复交叉设计包括:
Balaam 设计,即 4 序列 2 周期重复设计
序列 |
周期 Ⅰ |
周期 Ⅱ |
|---|---|---|
1 |
T |
T |
2 |
R |
R |
3 |
R |
T |
4 |
T |
R |
2 序列 3 周期重复设计
序列 |
周期 Ⅰ |
周期 Ⅱ |
周期 Ⅲ |
|---|---|---|---|
1 |
T |
R |
R |
2 |
R |
T |
T |
2 序列 4 周期重复设计
序列 |
周期 Ⅰ |
周期 Ⅱ |
周期 Ⅲ |
周期 Ⅳ |
|---|---|---|---|---|
1 |
T |
R |
T |
R |
2 |
R |
T |
R |
T |
1.3. 生物等效性研究设计要点#
生物等效性研究可用于化学药物仿制药的上市申请,也可用于已上市药物的变更(如新增规格、新增剂型、新的给药途径)申请。
受试者一般选择年龄为 18 周岁以上的健康受试者,且应涵盖一般人群的特征,包括年龄、性别等。
试验前需充分估计所需的样本量,以保证足够的检验效能。一般根据检验把握度及个体间变异大小来计算样本量估计值。若考虑在研究过程中可能因受试者脱落而导致样本量不足,在进行样本量估计时可考虑适当增加样本量。
针对仿制药的生物等效性研究中,应尽可能选择原研产品作为参比制剂,以保证仿制药质量与原研产品一致。
通常采用单次给药的给药方案,因为在单次给药过程中更易发现制剂药代动力学行为的差异。
一般情况下,应采用最高规格制剂一个单位(如单片或单粒)给药,且受试制剂与参比制剂的给药剂量应保持一致。
对于口服制剂通常需进行空腹和餐后生物等效性研究。空腹试验可发现制剂间的药动学差异;餐后试验则用于评价进食对生物利用度的影响。
各给药周期间应有足够长的清洗期(一般为受试制剂的 7 倍半衰期以上)。
一般使用以下药代动力学参数评价生物等效性:
实测药物峰浓度 \(C_{max}\)
从 0 时刻至最后一个浓度可准确测定的样品采集时间 t 的药物浓度-时间曲线下面积 \(AUC_{0-t}\)
从 0 时刻至无限时间的药物浓度-时间曲线下面积 \(AUC_{0-\infty}\)
一般用 非房室分析 方法估算药代动力学参数。可避免在房室模型分析中因为统计软件算法差异以及模型选择不同而导致的药代动力学参数计算结果不同。
1.4. 生物等效性数据处理与分析#
1.4.1. 统计假设#
用于分析生物等效性的数据集内通常应当包括至少一个周期且具有至少一个可评价的药代动力学参数。对于将进行分析的药代动力学参数,一般需要进行自然对数转换,以校正其数据分布的对称性。
生物等效要求受试制剂和参比制剂的药动学差异在一定可接受范围内,以 ABE 为例,其通过以下假设检验来进行统计推断:
原假设 \(H_0: \mu_T - \mu_R ≤ -\theta\) 或 \(\mu_T - \mu_R ≥ \theta\)
备择假设 \(H_1: -\theta < \mu_T - \mu_R < \theta\)
其中 \(\mu_T\) 与 \(\mu_R\) 分别为受试制剂与参比制剂对数转换后药动学参数的总体均值;\(\theta\) 为生物等效性界值,一般 \(\theta = ln(1.25)\)、\(-\theta = ln(0.8)\)。即受试制剂和参比制剂的几何均值比(公式如下所示)落在 80.00% — 125.00% 范围内才认为其生物等效。
备注
生物等效性界值与检验参比制剂百分比(percent of reference to detect)有关,一般为 20 %。记其为 \(PR\%\),则有:
一般的,我们使用 线性混合效应模型 来估算对数转换后药代动力学参数的最小二乘均值(least squares means,LS Mean)并以此作为对总体均值的估计值来进行假设检验。常使用双单侧 t 检验(two one-sided t-tests)或置信区间法进行假设检验。
1.4.2. 双单侧 t 检验#
双单侧 t 检验即对 \(\mu_T - \mu_R ≤ -\theta\) 与 \(\mu_T - \mu_R ≥ \theta\) 分别进行两次单侧 t 检验,对应的统计量分别为 \(t_1\) 与 \(t_2\):
其中 \(\bar{L}_T\) 与 \(\bar{L}_R\) 分别为受试制剂与参比制剂对数转换后药代动力学参数的 最小二乘均值;\(SE(MeanDiff)\) 为 最小二乘均值差 的标准误;\(N\) 为样本量。
当假设检验显著性水平为 \(\alpha\) 时,若 \(t_1 > t_{1-\alpha, N-2}\) 与 \(t_2 < -t_{1-\alpha, N-2}\) 同时成立,则拒绝原假设,认为两种制剂生物等效。
也可以计算得到最小二乘均值差的置信水平为 \((1-\alpha) \times 100\%\) 的置信区间为:
若上述区间落于 \((-20\%, 25\%)\) 内,则也可以认为两种制剂生物等效。
双单侧 t 检验的把握度(power)由以下公式给出:
其中 \(probt\) 为非中心 t 分布(non-central t-distribution)的分布函数,\(t_1\) 与 \(t_2\) 分别作为其非中心参数。利用此公式也可以计算得到给定把握度时的样本量。
1.5. 高变异药物与窄治疗指数药物的等效性分析#
某些药物由于生物利用度较低、酸不稳定、吸收前的广泛代谢等原因,导致其药动学参数的个体内变异(CV%)较大(≥ 30%),这样的药物我们称之为高变异药物(highly variable drug)。例如黄体酮、缬沙坦氨氯地平等均属于此类药物。
由于药动学参数的变异较大,此时若采用常规样本量和等效性判定标准,则可能会出现即使参比制剂与自身相比也无法证明其生物等效的情况。因此 CDE 与 FDA 推荐通过重复交叉设计试验,采用参比制剂标度的平均生物等效性(reference-scaled average bioequivalence,RSABE)方法,将等效性判定标准在 80% - 125% 的基础上适当放宽,可减少不必要的人群暴露,并达到科学评价不同制剂是否生物等效的目的。
RSABE 分析的步骤可概括如下:
计算参比制剂个体标准差 \(s_{WR}^2\)。
计算 \((\bar{Y}_T - \bar{Y}_R)^2 - \theta s_{WR}^2\) 的 90% 置信区间上限。
同时满足以下条件时可以认为两种制剂生物等效:
\((\bar{Y}_T - \bar{Y}_R)^2 - \theta s_{WR}^2\) 的 90% 置信区间上限小于等于 0。
受试制剂与参比制剂几何最小二乘均值比点估计值 90% 置信区间落于等效性范围内。
上述步骤中,\(\bar{Y}_T\) 与 \(\bar{Y}_R\) 为受试制剂与参比制剂对数药动学参数均值。\(\theta = \left(\frac{ln(\frac{1}{1 - PR})}{\sigma_{W0}}\right)^2\),其中 \(PR\) 为检验参比制剂百分比,一般取 20%;\(\sigma_{W0}\) 为法规限度(regulatory limit),一般取 0.25。
参考文献
90% 置信区间上限使用 Howe 近似算法得到:
Howe, W. G. . (1974). Approximate confidence limits on the mean of x + y where x and y are two tabled independent random variables. Journal of the American Statistical Association, 69 (347), 789-794.
窄治疗指数(Narrow therapeutic index,NTI)药物或窄治疗窗药物一般是指剂量或血药浓度的微小变化即可能导致治疗失败和/或严重不良反应,进而危及生命,或者导致永久或严重的残疾或功能丧失的药物。常见的此类药物有例如华法林、他克莫司等。
这些药物有效剂量与中毒剂量(或有效浓度与中毒浓度)接近;且血药浓度低于有效浓度时可能导致治疗失败,高于有效浓度则可能导致严重的不良反应。与一般化学药物相比,窄治疗指数药物进行生物等效性评价时,应采用更严格的等效性判定标准,以保证有效性和安全性。
窄治疗指数药物的生物等效性研究一般采用两序列四周期的完全重复交叉设计(即 T-R-T-R / R-T-R-T),以获得参比制剂和受试制剂的个体内变异。同时对药动学参数结果采用 RSABE 的方法进行分析。需要注意的是,对于窄治疗指数药物,在判断两种药物的生物等效性时除上文提及的两种条件外,需要额外满足以下条件:
受试制剂与参比制剂个体标准差比值 \(s_{WT}/s_{WR}\) 90% 置信区间上限小于等于 2.5。
只有当三种条件均满足时,才能认为受试制剂与参比制剂生物等效。
RSABE 相关详细的分析流程与统计算法可见 RSABE 分析流程与计算方法 章节。
备注
高变异药物与窄治疗指数药物的生物等效性分析可参考的相关指南文件: